文章标题:Machine Learning K-Means Clustering of Interpolative Separable Density Fitting Algorithm for Accurate and Efficient Cubic-Scaling Exact Exchange Plus Random Phase Approximation within Plane Waves
通讯作者:张振林,银栖麟,胡伟*,杨金龙*
背景介绍
作为当今材料计算方法中的主流方法之一,密度泛函理论(DFT)因其准确性和广泛的通用性而闻名。在DFT理论中,材料的电子结构和性质是电子密度的泛函,其中对计算结果准确性起决定性作用的是交换关联泛函,对交换关联泛函的探索被Perdew 在他的“雅各布天梯”(Jacob’s ladder) 中分为五类。
图1 雅各布天梯
其中的第五阶泛函,是目前最高等级的泛函,包括双杂化泛函(Double Hybrid functional),精确交换+RPA关联能(EXX+RPA)等,EXX+RPA方法能够准确捕捉系统中的关联效应,更好的描述长程范德华作用,这对于理解材料性质(特别是弱相互作用的材料)至关重要。
然而,EXX+RPA的计算量与电子数目的四次方成正比,计算标度为O(N4)。在平面波基组下,EXX的计算标度为O(N4)或O(N3),RPA的计算标度为O(N4),而且具有很大的指前因子,这昂贵的计算代价使得EXX+RPA一直以来只用于小规模的电子结构计算。
研究亮点
尽管之前有一些三次标度的算法被提出,如使用柯西积分解耦与ISDF算法结合,但其巨大的指前因子仍然使得大规模计算难以实现,因此探索一种新的计算RPA关联能的算法势在必行。
为了克服传统ISDF算法的限制,本文引入了一种增强的机器学习K-means方法。作者创新性地提出了一种经验权重函数“SSM+”,该权重函数可更精确地选择插值点,因此能够更准确地捕捉不同系统中的物理信息。这种机器学习方法计算插值点的标度为准二次,有效替代了基于平面波的EXX+RPA计算中昂贵的三次标度QRCP算法。该方法可以将RPA的计算标度从3.39降低到2.22,将EXX的总体计算标度从2.74降低到2.10。
作者使用基于MATLAB平台的第一性原理计算软件KSSOLV开发,可以使用MATLAB集成的GPU工具包来加速计算,实现了高达35倍的GPU加速。对于CPU计算时间,标准的四次标度方法需要22小时来计算Si128,使用K-means算法可将时间缩短到800秒,与标准四次标度算法相比,实现了高达100倍的加速。
通过这个改进的算法,作者对G2分子测试集的一些分子进行了测试,得出了与ABINIT高度一致的计算结果;作者成功计算了H2的解离曲线,给出了解离极限时曲线趋于零的正确趋势;最后,作者使用该方法正确预测了C18分子的平衡几何结构,与CCSD的计算结果一致。
数值验证
(1)为了研究计算体系和算法参数对精度的影响,作者选取了四个对称性不同的分子,选用不同的K-means经验权重函数:SSM、PSM、SSM+(本文),逐渐增大秩参数观察RPA关联能的收敛趋势以及绝对平均误差(MAE)变化情况。如下图所示,结果表明,随着秩参数(辅助基组数量Naux与秩参数成正比)增大,RPA关联能逐渐趋近于参考值Eref。SSM因为不能准确捕捉占据态信息,表现最差,几乎不能收敛;PSM可能会出现过拟合的情况,在某一秩参数之上误差陡然增大;而SSM+赋予占据态和未占据态相等的权重,随着秩参数的增大其计算精度表现出稳定和连续的收敛性质。
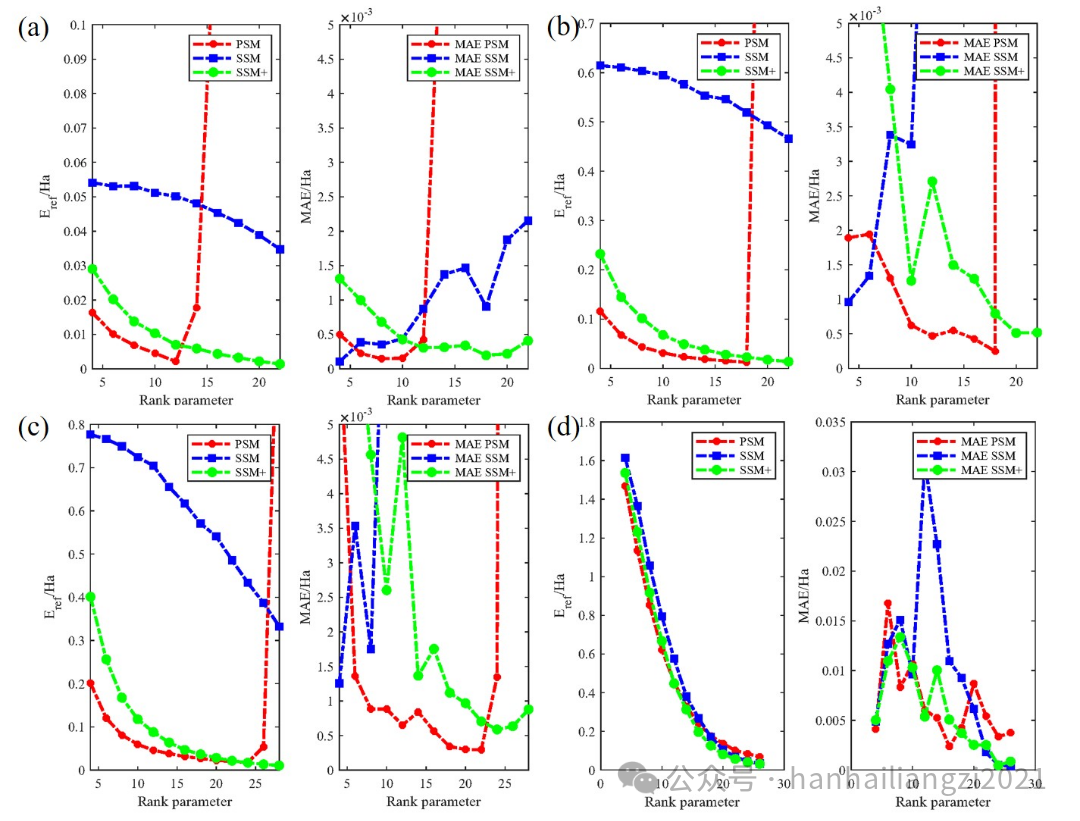
图2 四种对称性不同体系的权重函数测试 (a) H2 (b) CO (c) CH3OH (d) Si8
(2)为了测试本文算法的性能表现,作者使用三种不同的算法:标准算法、QRCP计算插值点、K-means计算插值点对不同大小的单Г点fcc硅体系进行了单线程测试(Si8-Si128)。对计算时间的标度拟合表明,对于EXX,ISDF(QRCP)计算标度与标准算法持平,但指前因子大于标准算法,ISDF(K-means)标度为2.10,小于标准算法2.74,但指前因子大于标准算法,其优势在大体系时才会体现出来。对于RPA计算, ISDF(QRCP)和ISDF(K-means)的拟合标度分别为2.56和2.22,而且指前因子比标准算法低。ISDF(K-means)大大降低了RPA的计算量,对于Si128这种大体系的RPA关联能计算,标准算法计算时间为79329s,ISDF(QRCP)计算时间为4156s,而ISDF(K-means)计算时间为800s,比标准算法快了近100倍。
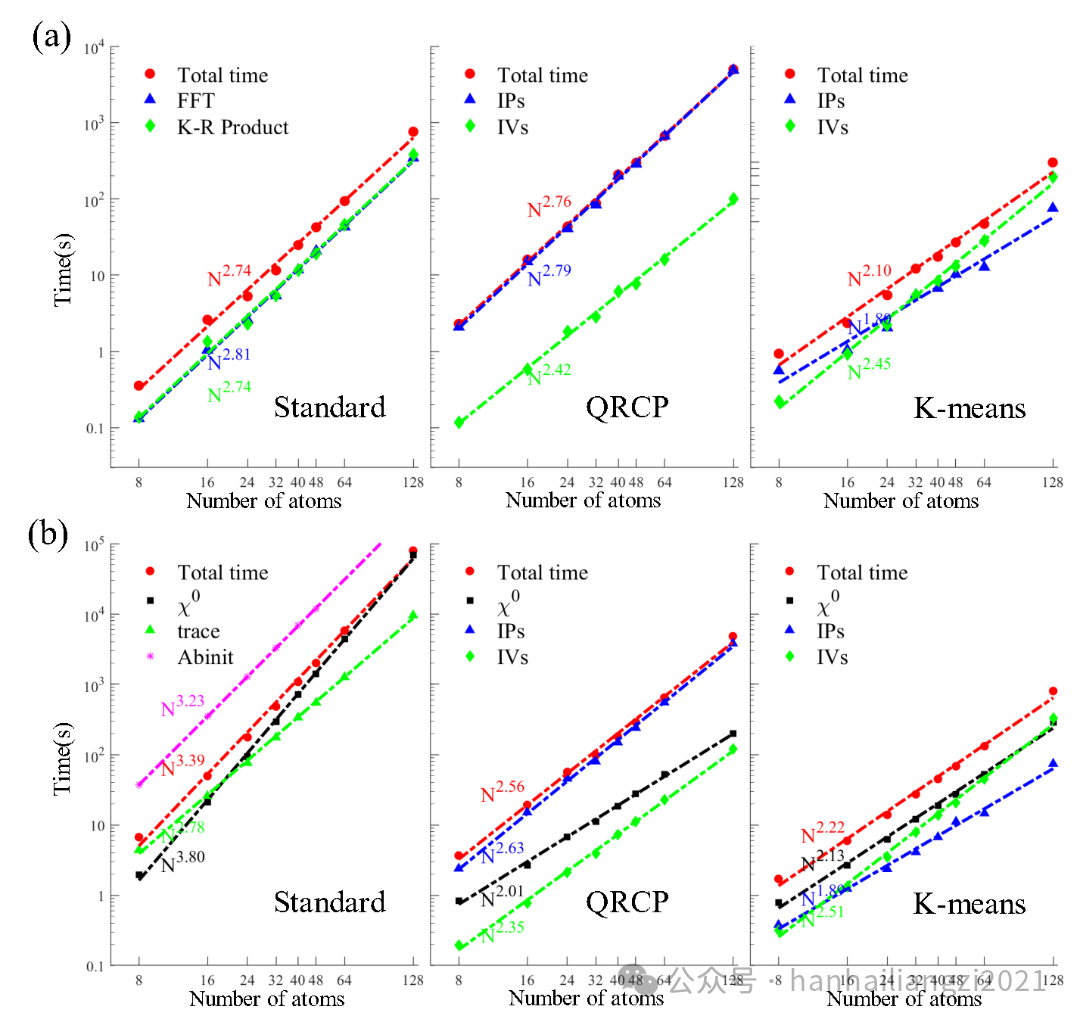
图3 EXX与RPA单线程计算时间随原子数目增大的变化 (a) EXX (b) RPA
(3)作者横向对比了ABINIT和Quantum Espresso的RPA计算功能。ABINIT目前只有标准算法(四次标度),Quantum Espresso使用cholesky分解来计算ISDF的插值点(三次标度)。计算体系仍然是fcc体相硅。结果表明,由于使用了K-means算法,KSSOLV的计算速度相比以上两款软件有了显著的提升。
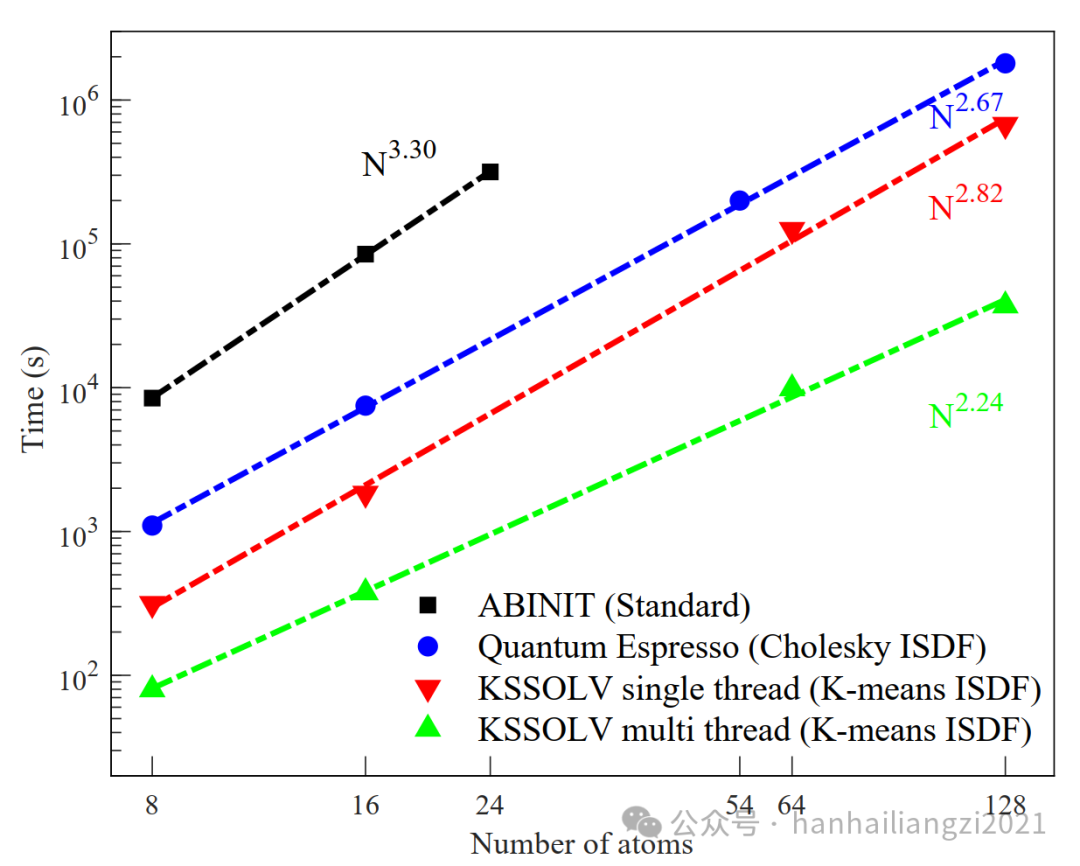
图4 不同软件的RPA计算时间对比 ABINIT、Quantum Espresso、KSSOLV
(4)作者测试了MATLAB中集成的GPU加速功能对算法的加速表现。如下图所示,GPU对于三种算法都有着不同程度的加速效果。其中QRCP加速效果最明显,对于Si48的RPA计算能加速18倍,对于K-means的加速比则能达到5倍。
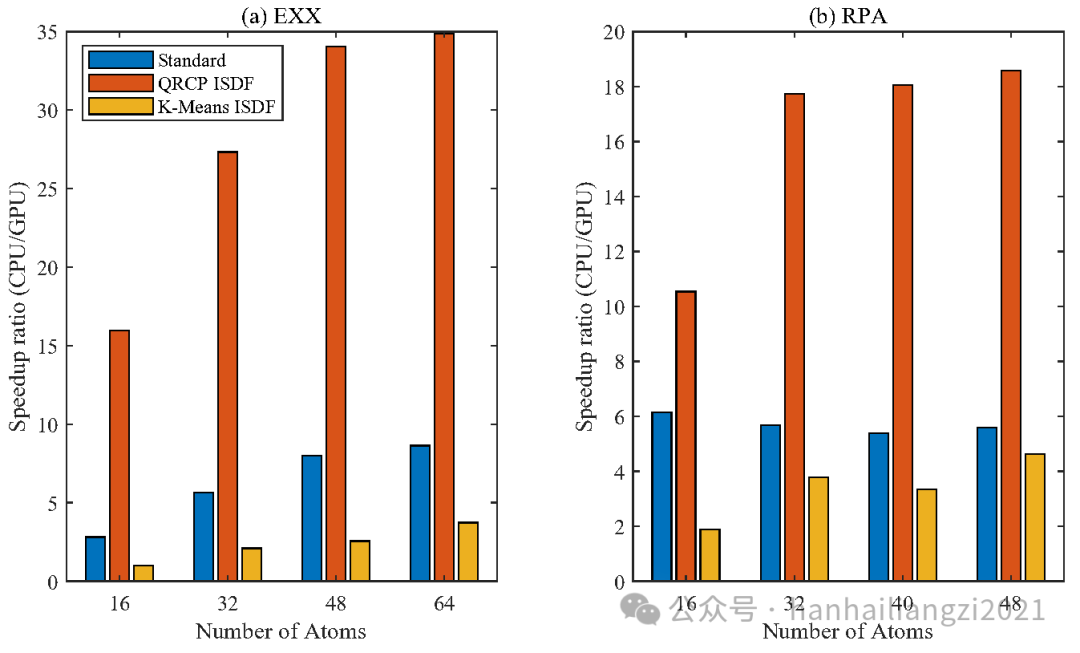
图5 GPU加速比 (a) EXX (b) RPA
算法应用
(1)作者基于KSSOLV 中实现的EXX+RPA功能计算了H2的解离曲线。如下图所示,GGA-PBE计算的原子化能通常会在解离极限处高估2eV,而使用EXX+RPA框架能够准确描述原子化能在解离极限处趋于零的趋势。
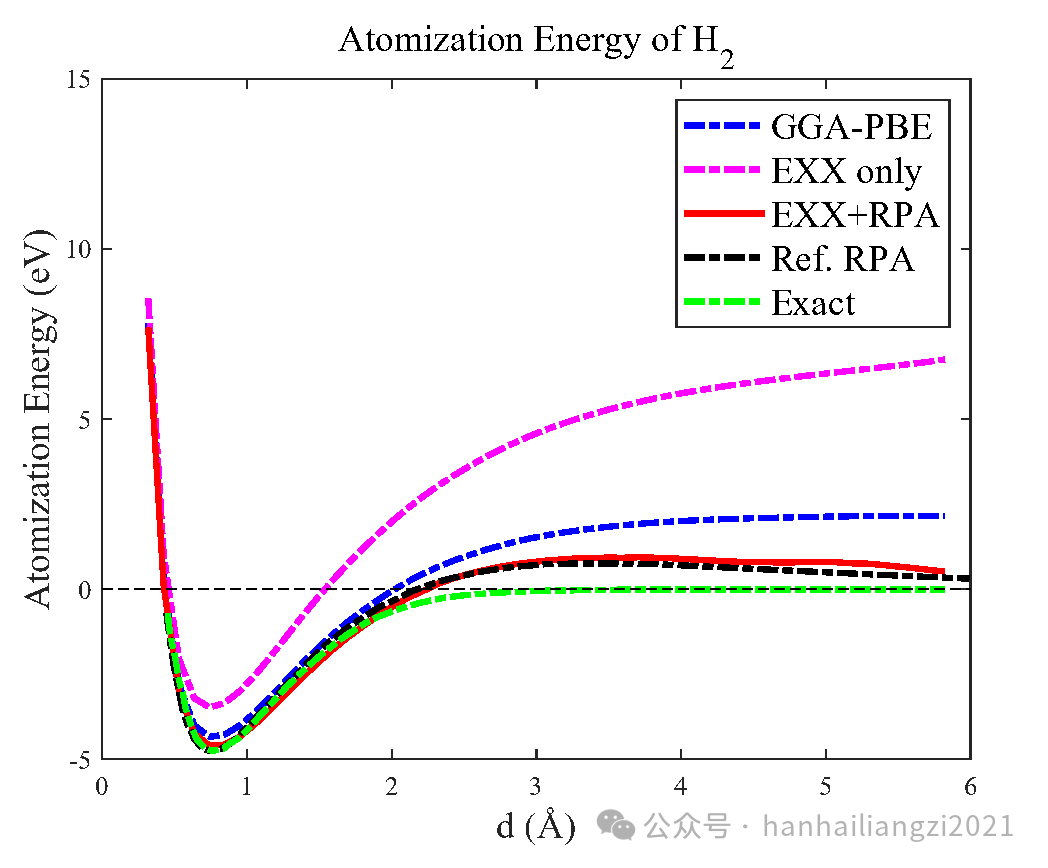
图6 H2的解离曲线
(2)作者基于KSSOLV的三次标度的EXX+RPA功能计算了C18的平衡构型。C18有两种可能的结构,一种是所有键全同,如图7上图的θ1=20°,一种是键长交替变化,即θ2≠20°。一般的DFT和微扰理论(MP2)预测出的是键全同的结构,而实验以及CCSD计算则表明真实结构是键长交替变化的。

图7 (上图)C18分子结构示意图 (下图)GGA-PBE, EXX+RPA, HXX+RPA, EXX only,
和 CCSD计算出的C18分子势能曲线对比
通过改变θ角(θ取值范围[0°,40°]),可以将计算出的总能量对θ=20°的总能量相对值Eref画出一条曲线,若Eref在θ=20°有极小值,则对应于键全同的结构;若Eref的极小值不在θ=20°,则对应于键长交替变化结构。计算结果表明,GGA-PBE预测的结构为键全同,而EXX only(只考虑EXX,不计入RPA关联能)、EXX+RPA、HXX+RPA(用HSE轨道计算EXX,用PBE轨道计算RPA)预测结构都是键长交替变化的,与CCSD一致。注意到EXX+RPA曲线的低谷相较CCSD曲线不明显,作者将其归因于EXX+RPA缺乏单激发贡献,所以计算不准确。通过使用HSE轨道计算EXX则可得到与CCSD更为接近的结果,即图中黑色曲线 。
了解KSSOLV软件
KSSOLV(Kohn-Sham Solver)是一款基于平面波基组的求解Kohn-Sham密度泛函理论的代码,利用MATLAB编程和计算,使用解释性语言,无需额外编译安装,支持Windows、Mac OS X和Linux系统,同时支持英伟达GPU加速,能够在个人PC端达到主流计算软件的计算速度。目前KSSOLV中已经实现了分子和固体的结构优化,电子能带结构计算(包括自旋轨道耦合效应SOC),局域密度近似(LDA)、广义梯度近似(GGA)和杂化泛函(HSE06)的交换关联泛函以及激发态电子结构(LR-TDDFT和GW)计算和动力学模拟(RT-TDDFT)。KSSOLV还可以用于验证公式和算法的正确性,其中已经实现的三种加速算法,包括自适应压缩交换算子(ACE)方法、投影的C-DIIS(PC-DIIS)方法和插值可分密度近似(ISDF)方法,加速了杂化泛函、激发态电子结构计算和动力学模拟,有希望实现针对数百原子中等尺度的分子固体材料体系激发态光电性质的计算模拟。关于KSSOLV软件的最新发布文章参见
Computer Physics Communications 279 (2022) 108424。
文章链接:
https://pubs.acs.org/doi/10.1021/acs.jctc.3c01157
